在上一章推文中,我们通过列举出细胞群的经典基因,然后通过umap图可视化后观察在哪个群集中表达,从而将那个群注释为我们认为的细胞名称。
相反,我们可以计算每个簇的标记基因,然后查找是否可以将这些标记基因与任何已知的生物学联系起来,例如细胞类型和/或状态。对于簇的标记基因计算,Wilcoxon 秩和检验等简单方法被认为表现最佳 。重要的是,由于集群的定义基于与这些统计测试相同的数据,因此这些测试的 p 值将被夸大
代码展示
首先我们先计算一下不同细胞群的marker基因:
sc.tl.rank_genes_groups( adata, groupby="leiden_res0_25", method="wilcoxon", key_added="dea_leiden_res0_25")
我们解释一下这段代码:
●groupby="leiden_res0_25":指定分组变量,leiden_res0_25 是 Leiden 聚类的结果列,用于将细胞分为不同的群体。这里的聚类分辨率为 0.25。
●method="wilcoxon":选择差异表达分析的方法为 Wilcoxon 秩和检验,这是一种非参数检验方法,用来比较不同群体之间基因表达的差异。
●key_added="dea_leiden_res0_25":指定将差异表达分析的结果存储在 adata 对象的 .uns 中,键名为 dea_leiden_res0_25。
接下来我们用气泡图展示一下前五基因:
sc.pl.rank_genes_groups_dotplot( adata, groupby="leiden_2", standard_scale="var", n_genes=5, key="dea_leiden_2")
●standard_scale="var":
这里的 var 表示按基因进行标准化,即每个基因的表达量会被减去该基因的均值,再除以该基因的标准差。这样,经过标准化后的每个基因的表达数据将具有均值为0,标准差为1。每个基因的表达量会被标准化,以消除不同基因表达水平上的差异。这样可以避免一些基因的表达量特别高或低,导致在后续分析中产生偏差。例如,如果某些基因的表达量远高于其他基因,这些基因的变化可能会主导整个分析过程。而通过标准化处理后,所有基因都会在相同的尺度上进行比较,减少了这种偏差的影响。

气泡图结果
如上图所示,很多差异表达的基因在多个簇中高度表达。我们可以过滤差异表达基因,以选择更多的簇特异性差异表达基因:
sc.tl.filter_rank_genes_groups( adata, min_in_group_fraction=0.2, max_out_group_fraction=0.2, key="dea_leiden_res0_25", key_added="dea_leiden_res0_25_filtered",)
●min_in_group_fraction=0.2:筛选时,只有当某基因在至少 20% 的细胞中在某个群体里有表达时,才认为这个基因在该群体中具有生物学意义。如果该基因在某个群体中的表达低于 20%,则会被去除。
●max_out_group_fraction=0.2:如果某个基因在其他群体中的表达超过 20%,则认为这个基因的表达可能是“非特异性”的,容易受到其他群体的影响,因此也会被去除。
接下来咋们可视化:
sc.pl.rank_genes_groups_dotplot( adata, groupby="leiden_res0_25", standard_scale="var", n_genes=5, key="dea_leiden_res0_25_filtered",)
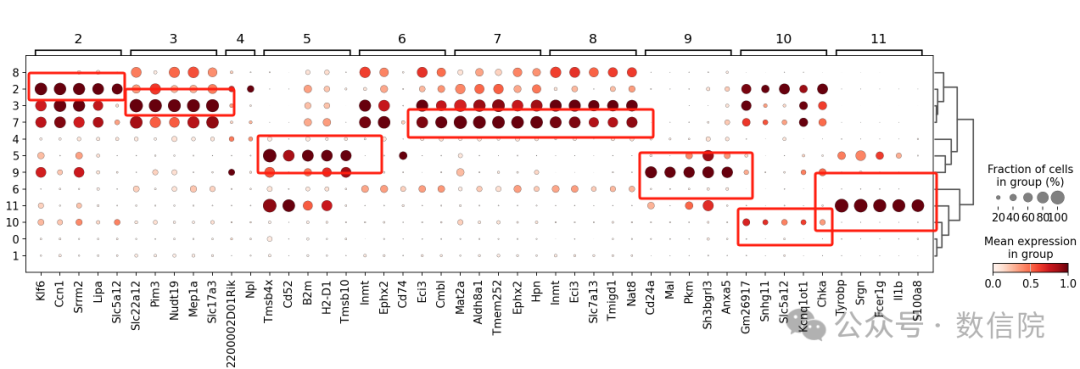
结果气泡图
可以看到,通过这一步,其中几个细胞群已经被标记有明显的基因,而有一些细胞群差异基因仍然不是很清楚。往往这种时候需要结合手动注释不断调整。
让我们看一下聚类11:
sc.pl.umap( adata, color=["Tyrobp", "Srgn", "Fcer1g", "Il1b", "leiden_res0_25"], vmax="p99", legend_loc="on data", frameon=False, cmap="Reds",)

结果图
可以看到,正好是在5和11两个细胞群中高表达。我们再继续看这几个基因,进行gpt搜索也可以初步确定其与单核细胞还有巨噬细胞相关。说明5,11两个细胞群实际属于巨噬细胞。