:HER理论计算VolmerHeyrovskyTafel计算以为基础,结合、,软件如等助力研究。经典案例揭示石墨烯缺陷的活性调控,前沿聚焦多尺度模拟、机器学习及界面效应,推动理性设计。
电催化氢气析出反应()的理论计算以量子力学方法为基础,通过构建反应路径的热力学与动力学模型,揭示催化剂活性起源并指导高效材料设计核心逻辑围绕反应机理、关键描述符及电子结构调控的关联展开。
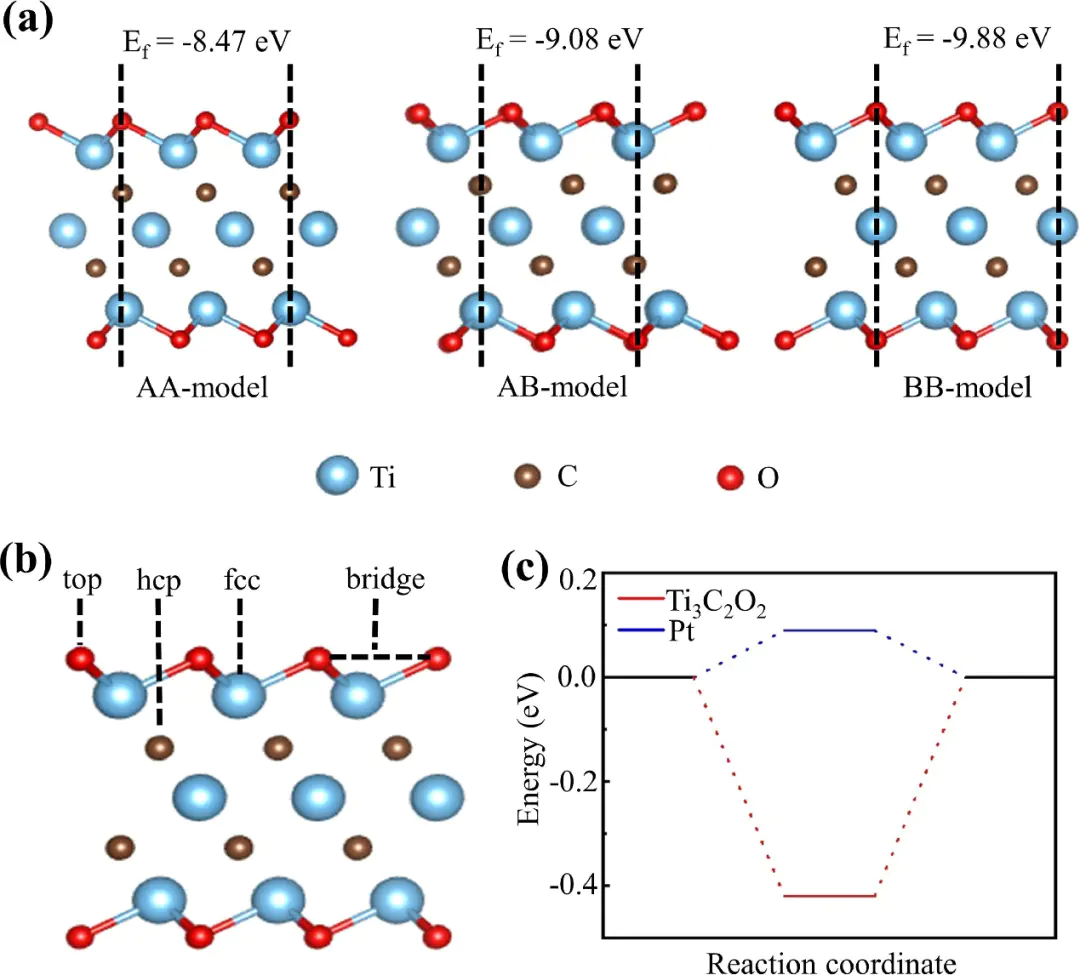
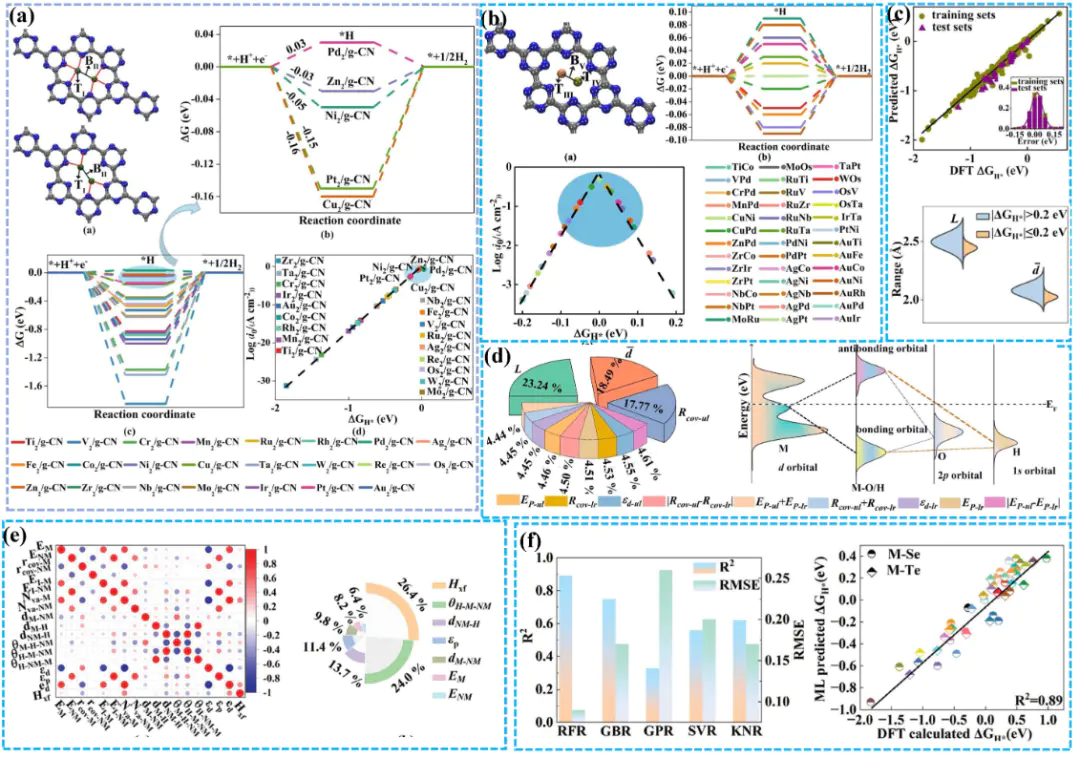
反应机理HER,电子与质子结合形成吸附态氢原子;步骤为电化学脱附,吸附态氢与质子、电子结合生成氢气;Tafel步骤为化学脱附这三步反应的速率差异决定了整体反应的主导机制,可通过斜率量化——当斜率b≈116 mV/dec时,Volmer步骤为决速步,表明质子吸附困难;当b≈30 mV/dec时,Tafel步骤主导,说明氢原子复合脱附是限速环节;而b≈40 mV/dec则对应Heyrovsky步骤主导,反映电化学脱附的动力学阻力。
关键描述符是关联理论计算与催化活性的桥梁,其中是最核心的热力学指标,HH*过电位(η)电子结构调控是优化描述符的核心手段,H类似地,过渡金属掺杂可通过改变带中心位置调控吸附能:d带中心上移会增强与H,下移则减弱吸附,这种“结构–电子态–吸附能” 的关联模型,为理性设计HER催化剂提供了原子级指导。
HER怎么算?
HER核心计算框架以密度泛函理论为基础,通过求解方程获得催化剂表面的电子密度分布,进而计算H吸附能、过渡态能垒及电子结构参数DFT的关键在于泛函与模型的选择:交换关联泛函多采用PBE-GGA,其在平衡计算效率与表面反应描述精度上表现优异,对于弱相互作用显著的体系(如层状材料),需添加色散校正以修正范德华力低估;溶剂化效应的处理则根据体系复杂度选择隐式模型或显式水分子层。
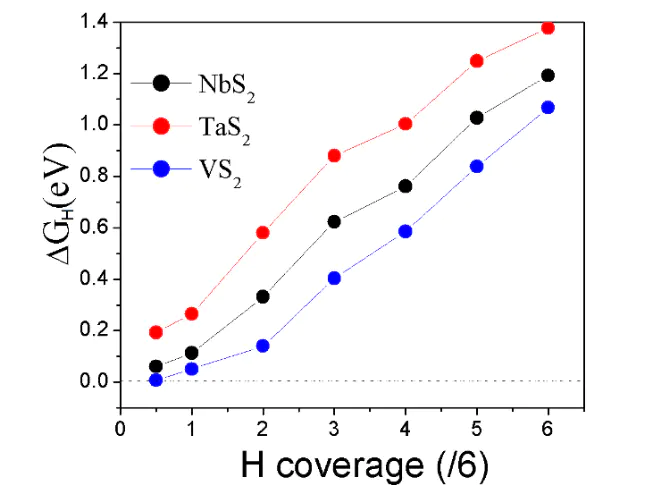
微动力学模型DFT在分析MoS₂的HER机制时,微动力学模型可量化Volmer-Heyrovsky与Volmer-Tafel路径的贡献比,发现边缘位点因Tafel步骤能垒低,更倾向于化学脱附,而基面则以Heyrovsky步骤为主。
在计算中引入电极电势,修正吸附能与反应能垒,该方法可模拟电化学界面的电势效应,使过电位计算更贴近实验条件。
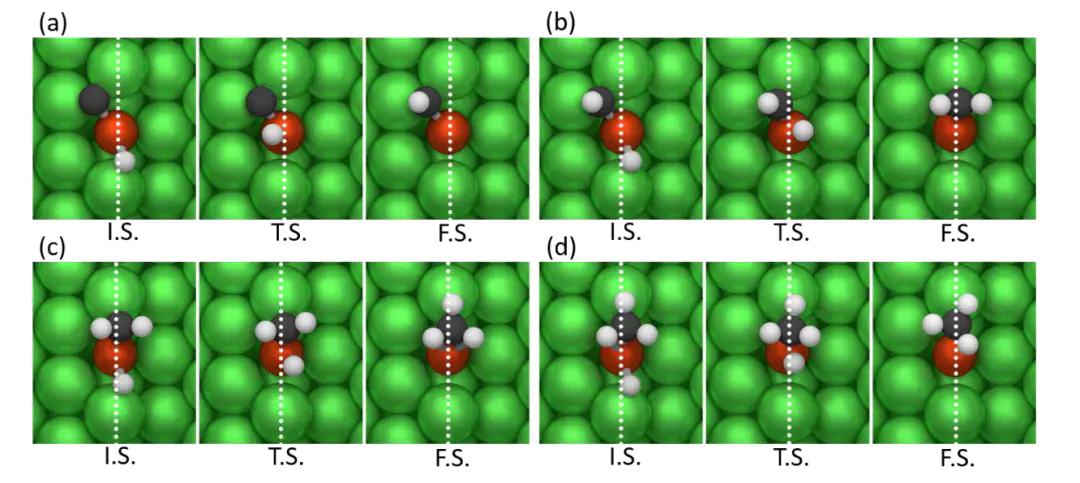
:采用平面波基组与投影缀加波赝势,擅长处理固体表面与界面体系,其Pulay DIIS算法可加速自洽场收敛,适用于缺陷、掺杂等复杂结构的电子结构计算,但需Linux环境编译,入门门槛较高。
Gaussian以分子轨道理论为基础,支持高精度方法,适合小分子团簇的反应路径模拟,但受限于基组规模,仅能处理原子的小体系;CP2K采用混合高斯/平面波基组,兼顾精度与效率,尤其擅长处理溶剂化体系,但对计算资源要求较高。
H催化剂的高效开发提供了新路径,其流程包括:首先构建材料数据库,通过DFT计算批量获取ΔG_H*、过电位等关键参数;随后以原子半径、电负性、d带中心等为输入特征,训练机器学习模型,建立结构与活性的映射关系;最终利用训练好的模型预测未计算材料的活性,筛选出潜在高活性体系。
但挑战在于实验环境的简化处理——现有模型多忽略温度、压力及动态界面效应,需引入主动学习策略,通过迭代补充高误差样本,提升模型对复杂实际条件的泛化能力。
缺陷调控HER活性
等人通过系统的理论计算,HER其研究设计从模型构建到机理分析形成了完整的逻辑链条。
HER吸附极弱DOI:10.1016/S1872-2067(21)63945-1
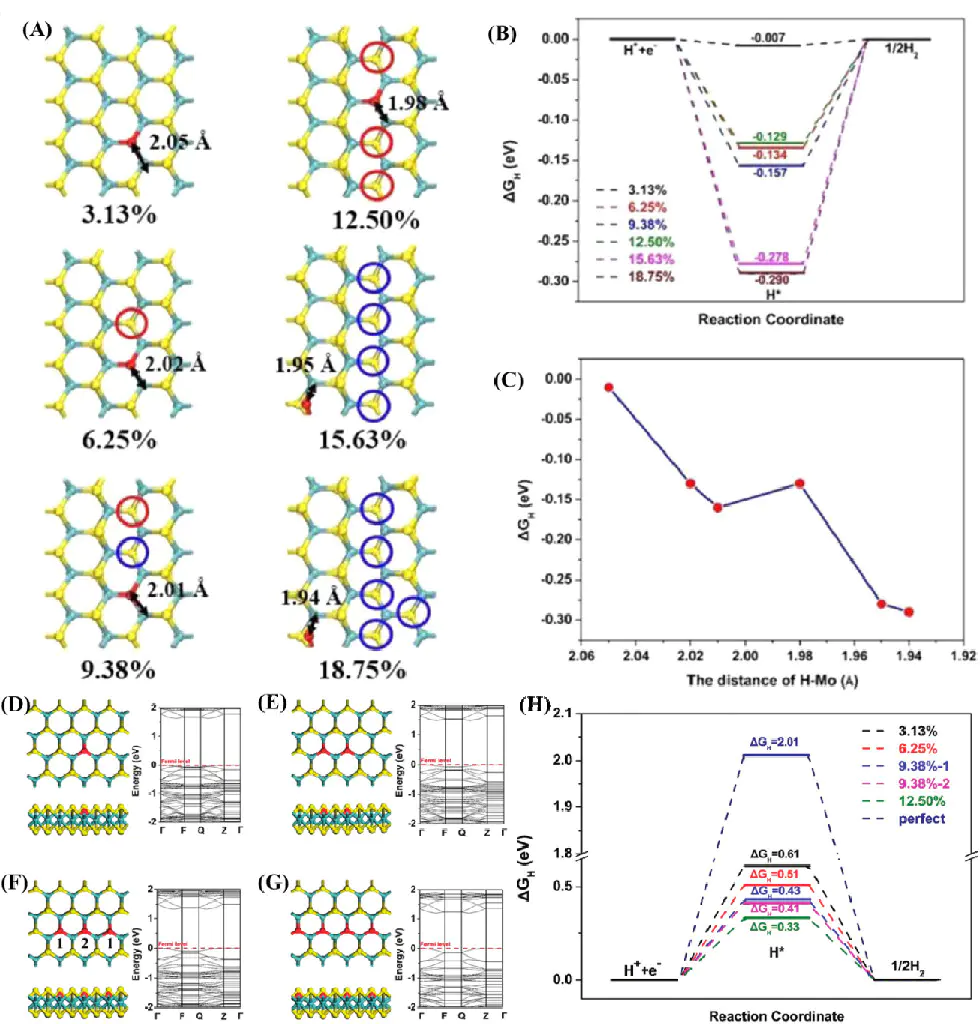
585反应路径模拟采用三态模型,计算各缺陷位点的ΔG_H*:结果显示585-3缺陷位点的ΔG_H*=-0.295 eV,7557-4位点的ΔG_H*=-0.187 eV,均显著优于本征石墨烯,其中7557-4位点的ΔG_HPt吸附强度。
了活性提升的微观机制:缺陷的引入使石墨烯的电子态密度在费米能级附近出现明显局域峰,这些局域态源于缺陷处碳原子的未成对电子——三配位C原子的p轨道未完全杂化,形成悬挂键,增强了与H电荷密度差分图显示,的轨道与缺陷C原子的p轨道发生显著电子云重叠,电荷转移量高于本征基面,证实缺陷通过增强电荷转移能力优化了吸附能。
过渡态7557研究结论明确:585和7557缺陷通过改变石墨烯的电子结构,使原本惰性的基面转化为高活性HER位点,其核心是缺陷诱导的局域态密度优化了H*吸附能与电荷转移效率。
该案例的价值HER前沿
HER多尺度模拟融合DFTDFT提供活性位点的吸附能、过渡态能垒等基础数据,KMC则基于这些数据模拟有限时间内的反应路径演化,考虑温度、压力、电解质浓度等实际条件的影响。
HER机器学习的应用面临“数解决这一问题需开发跨体系的通用描述符,将电子结构参数(如d带中心、Bader电荷)与宏观性质(如电负性、原子半径)结合,构建具有物理意义的特征空间。例如,基于“d带中心-ΔG_H*”的线性关系,可建立适用于合金、氧化物、碳材料的统一模型。
DOI:10.1002/celc.202400084
是提升复杂体系计算精度的关键,双功能催化剂的活性源于界面处的协同作用,需发展更精准的电荷分布算法。
QM例如,在Ni (OH)₂-Pt界面的HER模拟中,QM/MM方法揭示Li⁺通过静电作用稳定中间体,使Pt的ΔG_H-0.09 eV此外,(AIMD)可模拟界面水层的动态重构,捕捉H₃O⁺与活性位点的瞬时相互作用,发现传统静态模型忽略的质子传输捷径,使能垒计算误差降低0.15 eV。这些方法的发展为解析“催化剂–电解质–电极”复杂界面的HER机制提供了新工具。
氢吸附自由能与电子性质的内在关联,构建了从原子级结构到宏观催化活性的定量模型,为高效催化剂的理性设计提供了系统性蓝图。
的火山图关系明确了活性优化的目标(|≈0 eV当前研究仍需突破多重挑战:高通量计算平台的集成需解决软件兼容性与数据标准化问题,实现从材料数据库到活性预测的自动化流程;QM/MM捕捉动态电荷转移与溶剂化效应。