电场效应电子结构电双层量子模型与机制阐释界面作用,和模拟揭示电场降低活化能、诱导路径切换的微观过程,如反应中电场使能垒下降并引发机理转变,为催化调控提供外场干预新范式。
什么是电场效应?
电场效应是指外电场通过定向极化作用对催化体系电子结构产生的调控作用。
其路径的能量分布与中间体的化学行为,具体表现为以下三个维度的调控作用:在过渡态稳定化方面,外电场能够通过稳定极性过渡态显著降低反应活化能,当电场方向与反应偶极矩方向对齐时,活化能降低幅度可达以上,这一效应在酶催化体系中表现尤为突出——活性位点形成的高达1 V/nm的局部电场,可通过C=O键的Stark位移直接关联催化速率的提升,本质上是电场诱导的偶极–偶极相互作用优化了过渡态的电荷分布。
电子云分布在吸附能重构方面,电场通过调节催化剂表面的,改变反应中间体的吸附强度,DFT计算表明,+0.5 V/Å的电场可使Ni (111)表面*CO₂的吸附能增强0.8 eV,同时将CHO离解能垒降低0.3 eV,这一过程体现了电场对表面化学键能的动态调制。
催化及有机合成中的应用,为设计高效催化体系提供了通过外场调控电子结构的新范式,尤其是在电催化领域,电场效应与电极电位的协同作用,正成为优化反应路径与产物选择性的关键手段。
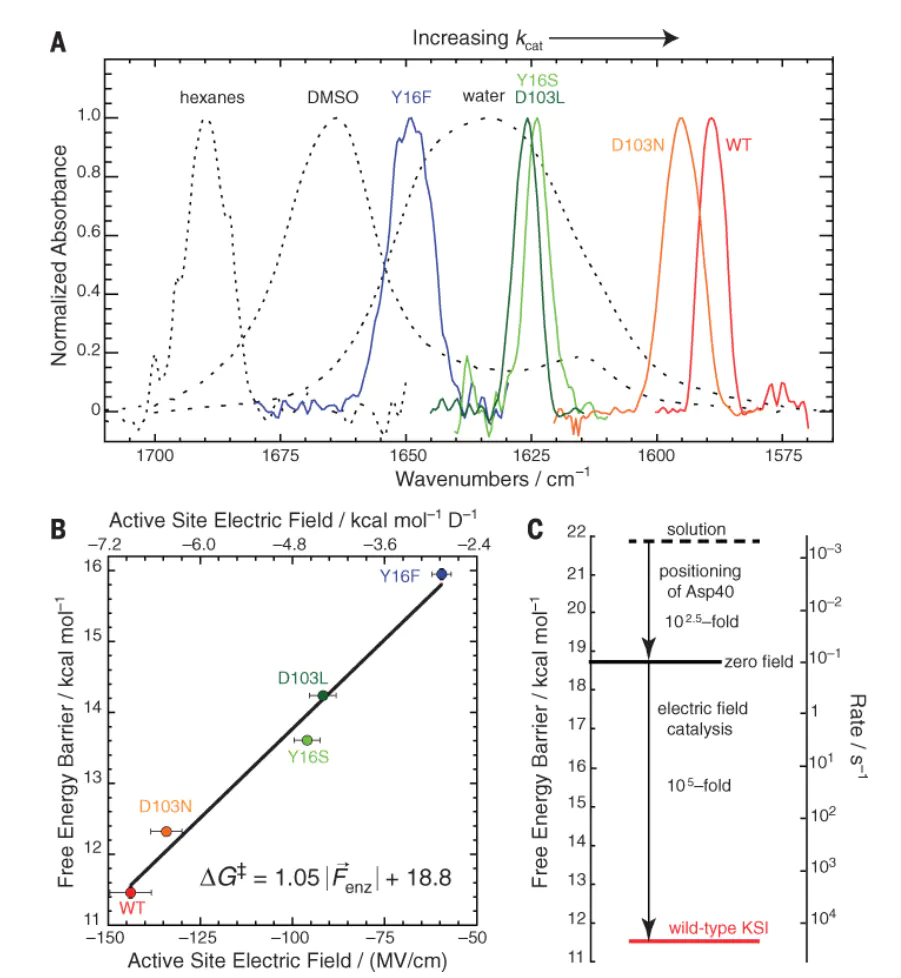
电双层量子模型
Discrete Helmholtz模型将电极/电解液界面描述为离散电荷层构该模型通过引入离散电荷的量子化效应,成功解释了振动和频光谱中观测到的电场诱导分子振动频率的偏移现象——当外电场作用于催化剂表面时,界面电荷层与吸附物种的偶极矩产生协同极化,导致化学键振动能级发生位移,进而影响反应中间体的吸附构型与活化能垒。
表面质子电流机制例如,在电催化析氢反应中,电场诱导的质子电流可加速H向活性位点的迁移速率,减少传质阻力,同时通过调节界面值优化氢吸附中间体的脱附能垒。
电场与催化剂相互作用调控的高效催化体系提供了关键理论依据。
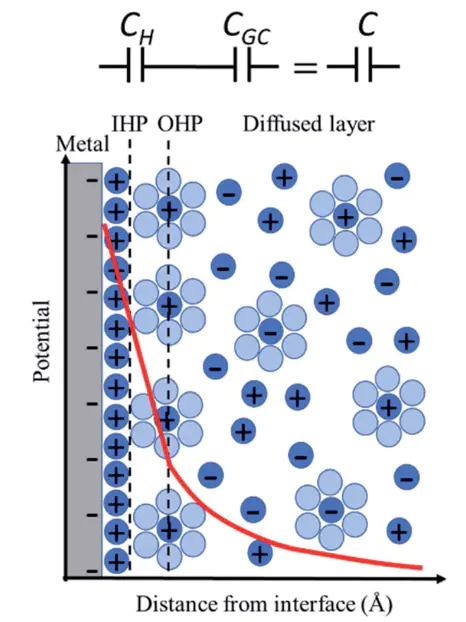
催化剂载体电荷转移
Pt功函数的下降本质上源于掺杂引入的载流子在电场作用下向Pt颗粒表面的定向迁移,导致面电子密度增加,费米能级附近的d轨道电子占据数改变。
该机制在电催化析氢反应中表现为:载体通过电荷转移调控Pt表面的电子结构,在不改变Pt本征晶体结构的前提下,通过界面电场效应实现催化活性的定向优化,为设计高效金属–氧化物复合催化剂提供了通过载体掺杂调控界面电荷转移的理论范例。
10.1103/PhysRevB.96.165405
电场效应模拟方法
在密度泛函理论框架下,电场效应的模拟方法主要通过电场嵌入计算与吸附能响应函数实现对催化体系电子结构与吸附行为的定量分析计算通过在哈密顿量中引入静电势项,直接耦合外电场与物质的电荷分布,从而精确描述电场对电子轨道能级与化学键强度的调制作用。
Ni该方法通过,实现了对催化活性位点电子结构的外场调控模拟,为揭示电场诱导的键活化机制提供了原子尺度的理论工具。
了吸附能变化与电场强度的定量关系。该函数表明,吸附能的变化由非线性的极化效应与线性的偶极电场相互作用共同决定:极化率越大,电场诱导的分子变形越显著,吸附能降低越明显;偶极矩分量则反映了电场方向与分子偶极取向的协同效应,当二者方向一致时,线性项贡献占优,可通过定向极化直接降低吸附能。
这两种模拟方法相辅相成:电场嵌入计算从第一性原理出发,精确刻画电场对电子结构与化学反应能垒的影响,适用于复杂催化体系的机理分析;吸附能响应函数则通过参数化模型,快速预测电场对吸附行为的定量调控,便于高通量筛选与催化剂设计。
二者共DFT提供了微观视角,更推动了 “电场调控催化” 从经验设计向理论指导的范式转变,在电催化、光电催化等领域具有重要的应用价值。
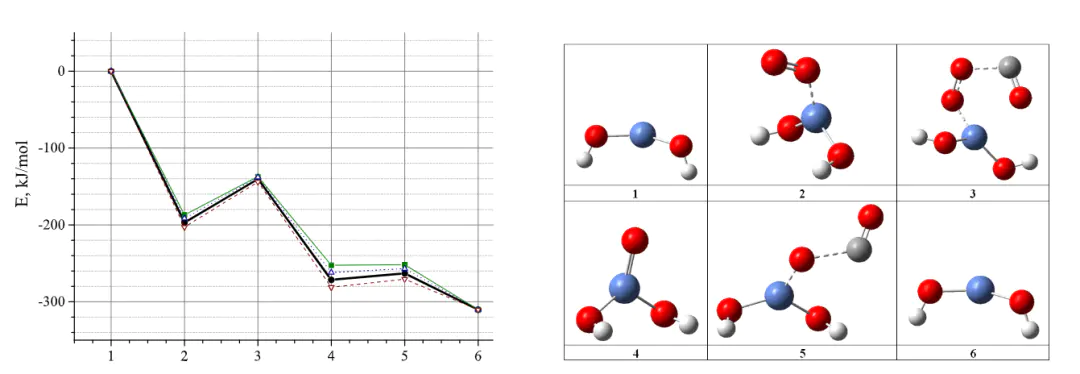
从头算分子动力学
AIMD在显式溶剂化模型中,对界面体系的模拟显示,施加电场后CO₂分子的O-C-O键角从173°扭曲至168°,这种结构畸变通过削弱O-C-O的线性共轭效应,显著降低了COOH中间体形成的能垒,揭示了电场通过调控反应物构型促进电催化CO₂还原的微观机制。
自由能AIMDDOI:10.1063/5.0066196
电场调控Diels-Alder反应
在电场调控有机反应的研究中,反应作为典型的环加成反应,其协同外场对化学反应路径的重构机制提供了重要范例。
以丁二烯与丙烯醛的反应体系为例,该研究选择沿反应轴施加的定向电场,通过密度泛函理论结合过渡态搜索,系统解析了电场对反应能垒、路径及电荷分布的调控效应。
协同机理即双烯体丁二烯与亲双烯体丙烯醛通过六元环过渡态同步形成两根σ键与一根π键,其活化能垒为80 kJ/mol,符合该类反应的典型热力学特征。
更关键的是,当电场强度超过0.4 V/Å时,发生本质转变——从协同环加成机制切换为分步离子机理,具体表现为首先发生Michael加成生成两性离子中间体(C⁺-O⁻),随后通过环化步骤形成最终产物。
电荷分析2.1 D这种 “离子化效应” 通过电场与分子偶极的定向耦合,诱导反应中间体从非极性向极性结构演化:在协同过渡态中,电场促使丁二烯的π电子云向丙烯醛的缺电子双键偏移,增强碳碳键形成过程中的电荷转移;而在分步机理的Michael加成步骤中,电场进一步稳定了碳正离子与氧负离子的电荷分离态,使该中间体的能量低于协同过渡态,从而主导反应路径。
电场强度下,电场主要通过稳定极性过渡态降低协同路径的能垒;当电场超过临界值时,极性中间体的能量优势导致反应路径切换,形成更高效的分步离子机理。
从理论层面看,该研究验证了电场作为“非共价催化剂” 的可行性:无需改变反应物化学结构,仅通过外场调控分子电荷分布,即可实现对反应路径与动力学的定向控制。
例如,电场诱导的两性离子中间体稳定化策略,可为设计高选择性环加成反应提供新思路,尤其是在合成具有特定立体化学结构的药物中间体时,通过调节电场参数有望精准控制产物构型。
电场效应值得注意的是,电场调控的非侵入性特点(无需引入外加试剂、可动态调节)使其在绿色化学与连续流反应中具有潜在应用价值,有望推动“电场催化” 从理论研究向实际合成工艺的转化。
Diels-Alder揭示了外场调控化学反应的普适性原理——通过定向极化作用诱导电荷重新分布,实现对反应路径、中间体稳定性及能垒的协同优化。
这一机制为拓展电场在有机催化、DOI:10.1039/C8CS00354H
总结
电场效应的理论研究正通过多维度计算策略实现深度拓展:–活性关联模型加速电场调控策略的优化设计;AIMD)模拟微秒级电场波动对质子耦合电子转移(PCET)过程的影响,阐明动态电场下催化界面的电荷传递与质子迁移协同规律。
这些研究共同证实电场作为 “第四维度调控手段” 的独特价值——其研究需聚焦多尺度模型的构建,以实现微观电子结构调控与宏观反应动力学的理论衔接,推动电场催化在能源转化与有机合成中的实际应用。